|
| Guess What! - ESTP Case 23 |
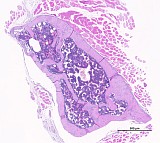
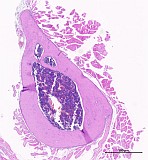
Mouse, male, 16-weeks study, found dead on test day 91, femur and tibia with joint (Fig. 1-3). Control slides from an unaffected male (Fig. 4-6).
| Click on the images below for a larger view. |
|
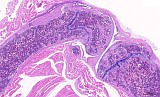
Fig. 1: Femur, tibia, longitudinal, H&E
|
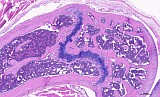
Fig. 4: Femur, tibia, longitudinal, control, H&E
|
|
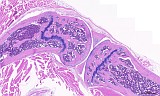
Fig. 2: Femur, longitudinal, H&E
|
Fig. 5: Femur, longitudinal, control, H&E
|
|
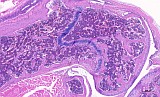
Fig. 3: Femur, transverse, H&E
|
Fig. 6: Femur, transverse, control, H&E
|
Morphologic Description
Figure 1-3 show the stifle of an oim/oim homozygous mouse: the femorotibial joint in longitudinal section (fig.1-2) and the femur in a transverse section (fig.3). Main finding is a decrease of cortical and trabecular bone in femur and tibia (osteopenia) with porous corticalis and extended medullary cavity. The formation of primary and secondary spongiosa in the subchondral plate is markedly reduced. Only mild regressive changes are visible in the joint cartilage, and the joint capsule shows slight reactive mononuclear synovialitis. Figures 4-6 illustrate the morphology in a wild type (+/+) control mouse.
Proposed Diagnosis
Osteogenesis imperfecta
Discussion
Eight contributions were received for this case.
Background of this lesion is a mutant mouse model (B6C3Fe a/a-Col1a2oim/J) developing osteogenesis imperfecta (OI) in homozygous offspring. Spontaneous mutation in the pro-alpha2 chain of type I collagen leads to osteopenia, cortical thinning ("brittle bone"), progressive skeletal deformities, fractures, and small body size. Type I collagen is found in connective tissue, cornea, dermis and tendon and is the most abundant collagen in the human body. So far, this mouse strain serves as research model for human osteogenesis imperfecta type I.
Histopathological feature of this OI model is hypoplasia of cortical and trabecular lamellar bone based on osteoblastic insufficiency to generate normal bone matrix and mineralization. Consequently the lamellar bone exhibits a thin and brittle phenotype, which can lead to deformation and fracture of long bones and compression of the spinal column. Reduced callus is often seen in fractured bones. Growth plates show rather normal composition at reduced height, but decreased spongiosa formation in the metaphysis. Periosteal lamellar bone formation is insufficient, while the endeostal resorption is not affected which can lead to thin, disorganized and porous cortical bone with extended Haversian canals.
The proposed diagnoses for this case included decreased bone/decreased thickness (of femur), osteoporosis, decrease or atrophy of knee joint cartilage, and chondrodysplasia.
Decreased bone/thickness correctly describes the bone morphology in our specimen which did not only affect the femur but also the tibia.
Osteogenesis imperfecta is assigned to the classification of hereditary osteoporosis, so far this diagnosis properly addresses the unequal or insufficient mineralization of bone matrix.
Chondrodysplasia or atrophy of the knee joint cartilage is not described as a primary entity in this OI model but can be expected as secondary change in deformed and incongruent joints.
Leukemia was received as additional diagnosis which could not be confirmed.
References
- The Jackson Laboratory Mouse Strain Datasheet - 001815 B6C3Fe a/a-Col1a2oim/J
https://www.jax.org/strain/001815
- Kamoun-Goldrat AS, Le Merrer MF (2007)
Animal models of osteogenesis imperfecta and related syndromes.
J Bone Miner Metab 25: 211-218
- Phillips CL et al. (2000) Oim mice exhibit altered femur and incisor mineral composition and decreased bone mineral density.
Bone 27(2): 219-226
|
|